|
| The view of Lasry the other day on my way into lab. |
A few weeks ago I took the GRE as part of my quest to continue graduate school in a Ph.D. program. Standardized testing is such a money scam given all of the fees involved from taking the exam to getting your scores and sending them to your schools. Furthermore, I don't think standardized exams are particularly informative, which is funny because Clark was just in the news because
they are no longer requiring SAT scores for undergraduate admissions.
But I digress: I'm not here to complain how ridiculous the whole process of taking the GRE is. I'm just super glad the GRE is behind me and I can focus on
things that actually matter. Yet ever since, I've been taking it slow with my workload because I needed a bit of a break. Thanksgiving marked the end of that break though, and now it's time to jump right back into things to finish up a productive semester.
I have a very hopeful list of things I'd like to accomplish by the end of the semester in mid December. But because I'll need to be working a lot more hours at the bookstore to help with the end of semester rush, it's going to be a big squeeze.
 |
| We're starting to get books for the spring semester at bookstore, & they're piling up. |
 |
| This picture is enhanced with the iPhone app Cat Effects. |
Meanwhile in the lab, I have
heaps to do. I want to finish up my experimental assays, screen and grow out more diatom cultures for future experiments, prepare a presentation and do some writing, and finally start with some
RT-PCR experiments.
Now previously
I extracted RNA from my frozen cell samples and used the mRNA present from the cell samples to generate cDNA.

RT-PCR, or real time PCR, will allow me to quantify the cDNA that I have made from my RNA. This is because RT-PCR can measure the number of DNA copies at each copy cycle by measuring the fluorescence of a special dye that hybridizes with the DNA. This is pretty snazzy, but it looks like I'm getting myself into a lot of grunt work.
You see, in order to get precise data from RT-PCR, one usually runs three replicates from one DNA sample and compares the output. But it's not like I only have one DNA sample. I have tons.
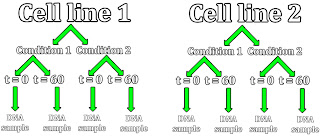
This is because I have 8 DNA samples at the minimum per experimental assay that I run in the beginning: I have two different cell lines that I test per assay, which get divided between two different test conditions, from which I take cell samples at multiple timepoints.
For the time being, I'm going to start with time zero and my end time of 60 minutes. This will hopefully give us start and end data that will display the overall trend of my experiments.
But because of
positional effects, I need to run more than one experimental assay. I'll probably run three sets of samples through RT-PCR, but I hope to have up to 5 samples completed and ready for RT-PCR by the end of next week.
 |
| This is from today while I waited for my next sampling time. |
Wait, what's that positional effects you just mentioned a second ago? Oh, yeah, positional effect. Because we transformed our diatoms with ballistics, our DNA was randomly inserted into the diatom's genome. By chance, the DNA we're trying to measure in our experiments might be inserted into a region that is either expressed more or less often, which would skew our data. By gathering data from multiple replicates, we'll get more accurate data regarding gene expression. Cool how science works, huh? We think of the neatest little caveats. Well, I mean, my professor does.
But sometimes I do too!
Like last month or so I came up with a solution as to why our cultures weren't growing so well. And then more recently I figured out how to
screen my diatoms for our reporter gene more effectively.
So yes, sometimes I come up with cool things too.
 |
| The Jonas Clark building. I can't stop taking pictures from this vantage point! Gah! I love it! 11/18/2011 |