
In re-examining my preliminary data on Wednesday, I realized I had collected some positive results! I had run a set of samples through a QRT-PCR reaction on Tuesday with the new primers that I received on Monday. These samples were from my experimental assays and represented different time points and test conditions. When I looked at the results on Tuesday, I didn't think much of them, other than the fact that my no template controls (the reactions that are supposed to be blanks) gave me some product, indicating the reactions are still not 100% clean.

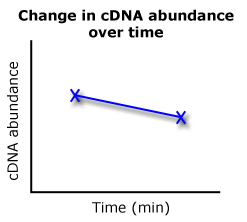
If sample A was the time 0 value and sample B was the time 60 value, we'd essentially get a graph like this on the right. Sample A's Ct of 25.88 is greater in cDNA abundance than sample B's 27.34 Ct. My QPCR comparisons will look a lot like these two cartoons, where I compare Ct values and determine what that data looks like on a graph.
So here I am going back through the data, comparing starting Ct values (0 minutes) with end Ct values (60 minutes) between two different treatments and DNA constructs from my transformed diatoms. Of these eight Ct values (four combinations, two time points each), I realized I had something. My three "controls" all showed the appropriate increase or decrease in mRNA transcript levels, mirroring previously established results by our lab. But when I looked at the experimental line of diatoms, the line I'm most interested, this pattern completely changed from its control. OHHHH YEAHHHH.
 |
| Positive preliminary results: victory! |
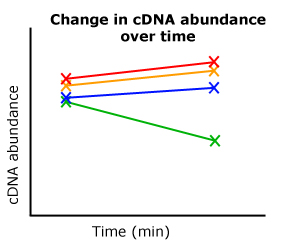 |
| A cartoon representing my results. |
However, I'm not out of the woods yet. I'm still having trouble with my primers and I may need to order the primers a third time. My no template controls are still giving me product, but that may be due to primer dimers. I'm going to run a gel in an attempt to see what is going on, but I may have to eventually sequence the bands to see whether I have some plasmid DNA contamination that I don't know about. All of this will be sorted out, hopefully, sooner or later. But I have some other projects going on that I really should take care of as well. Juggling so many different things is really time consuming, but that's a topic for another time.








{kind=link}
{kind=link}