A few weeks ago, I posted about staving off a massive culling of diatoms.
As of right now, my diatoms are still on the mend but I hope to have things all sorted out and lined up by the end of the week. To do this, I'll be checking on my plates and ensuring all of my diatom lines are accounted for before I clean out the culture room, which is in desperate need of being tidied up.
In the video below, I give my account of what it was like to prevent my diatoms from dying. Hopefully I will have been successful in doing so.

Showing posts with label genetic transformation. Show all posts
Showing posts with label genetic transformation. Show all posts
Sunday, January 29, 2012
Saturday, September 17, 2011
The Genetic Transformation of Diatoms with Inducible Expression Plasmids
Last month my undergraduate/now graduate research project at Clark University reached a new level of awesome with the transformation of diatom cells with plasmids I've been working on for several semesters. It's been a long time coming for sure, with both problems encountered in the lab project and juggling other college courses.
In my blog I've chronicled some steps along the way on my project, and I've talked about:
Now I am undergoing the process of selecting lines of transformed diatoms for the next step in our project. But we've recently realized that the light intensity in our growth chamber is much brighter than previously published experiments growing transformed cultures of diatoms. Increasing light intensity reduces the amount of chlorophyll made by plants and algae. Why is this important? Two reasons:
I've been growing diatoms in 3 mL cultures in little dishes, which you can see in the back left. Groups of 3 dishes sit in a petri dish to allow easier transportation. These dishes however require 3 mL of culture just to cover the entire bottom of the dish, which means the cells are going to be fairly diluted. I also started 1 mL cultures in the test tubes on the right, where porch screen blocks out a lot of the incoming light. Some of the 3 mL cultures also have screen on top of them to block out some of the light.
Aside from trying to grow our cells in liquid culture, I've been looking at them under a fluorescent microscope to try to determine whether our cells are fluorescing due to GFP expression or whether it's residual glow from chlorophyll. UV light from the microscope excites all pigments in the sample, and then I look at the sample through different filters that only allow certain wavelengths of light to show through. However, our current filter set up hasn't allowed a clear delineation between GFP and chlorophyll yet. Most of our pictures look something like this:
 ...but they usually have a lot more background color in there too.
...but they usually have a lot more background color in there too.
My next steps will be to either design or order commercial GFP GSPs--gene specific primers for GFP. That way we can run a PCR on some diatom DNA and determine whether they have the GFP gene they are supposed to be transformed with. Because I have a great control (the original plasmid DNA), I can test a whole bunch of diatom colonies at once and be able to select appropriate lines of diatoms faster.
In my blog I've chronicled some steps along the way on my project, and I've talked about:
- phase one of my project and phase two of my project and creating posters to present my project at campus-wide functions,
- transforming bacteria to clone pieces of plasmid and constructed plasmids,
- cutting pieces of DNA out of plasmids so they can be pasted into different plasmids,
- sequencing pieces of my plasmids,
- constructing my plasmids,
- completing my NiR plasmids
- and preparing for the diatom transformations.
Now I am undergoing the process of selecting lines of transformed diatoms for the next step in our project. But we've recently realized that the light intensity in our growth chamber is much brighter than previously published experiments growing transformed cultures of diatoms. Increasing light intensity reduces the amount of chlorophyll made by plants and algae. Why is this important? Two reasons:
- Less chlorophyll will reduce the pigmentation of our cells, making them harder to see once we transfer them to liquid culture.
- Our gene giving resistance to our antibiotic (that allows us to select for positive transformants) is driven by a chlorophyll-associated protein, which means expression may be reduced in higher light intensities. If chlorophyll expression is reduced by high light levels, then this may reduce the activity of the chlorophyll-associated protein that drives the antibiotic resistance in our diatoms, which means less resistance to the antibiotic in the media. This ultimately means reduced or no growth.
| Transformed diatoms grow on agar plates (left foreground) an in liquid media (left background and right). |
Aside from trying to grow our cells in liquid culture, I've been looking at them under a fluorescent microscope to try to determine whether our cells are fluorescing due to GFP expression or whether it's residual glow from chlorophyll. UV light from the microscope excites all pigments in the sample, and then I look at the sample through different filters that only allow certain wavelengths of light to show through. However, our current filter set up hasn't allowed a clear delineation between GFP and chlorophyll yet. Most of our pictures look something like this:

My next steps will be to either design or order commercial GFP GSPs--gene specific primers for GFP. That way we can run a PCR on some diatom DNA and determine whether they have the GFP gene they are supposed to be transformed with. Because I have a great control (the original plasmid DNA), I can test a whole bunch of diatom colonies at once and be able to select appropriate lines of diatoms faster.
Tuesday, September 13, 2011
Glowing Green Cells
Just before the weekend, my professor wanted to look under the microscope to see whether our transformed diatom cells were expressing our reporter gene GFP. I've been culturing several different lines of transformed diatoms since the transformation. These cells were co-transformed, which means the resistance plasmid is separate from the GFP plasmid--that is the transformation process shot two different plasmids at the diatom cultures. Therefore we need to select for diatom lines that posses both plasmids: the resistance plasmid and the GFP plasmid.
Below is a poster I made briefly describing each of the three main steps in the process: transformation, growing the diatoms on selective agar plates, and subculturing diatom cells in selective liquid cultures.

The diatoms will only grow if they received a copy of the resistance plasmid. And well, we can tell that the diatoms don't particularly like the antibiotic we put in the growth media:

By performing a simple antibiotic sensitivity test, we can show that diatoms will not survive in the presence of the antibiotic unless they receive the plasmid that confers resistance.
Below is the first round of pictures of cells expressing GFP. These pictures came from a single colony on an agar plate, which means I didn't transfer it to a liquid culture (i.e. it was sacrificed for science). However it does give us promise that we will have GFP-expressing lines of diatoms in the near future/ I've been working on growing up the cells in liquid cultures and hope to have something by the end of the week. After that, it's a matter of growing up larger liquid cultures before we can test some science!
Below is a poster I made briefly describing each of the three main steps in the process: transformation, growing the diatoms on selective agar plates, and subculturing diatom cells in selective liquid cultures.
The diatoms will only grow if they received a copy of the resistance plasmid. And well, we can tell that the diatoms don't particularly like the antibiotic we put in the growth media:

Below is the first round of pictures of cells expressing GFP. These pictures came from a single colony on an agar plate, which means I didn't transfer it to a liquid culture (i.e. it was sacrificed for science). However it does give us promise that we will have GFP-expressing lines of diatoms in the near future/ I've been working on growing up the cells in liquid cultures and hope to have something by the end of the week. After that, it's a matter of growing up larger liquid cultures before we can test some science!
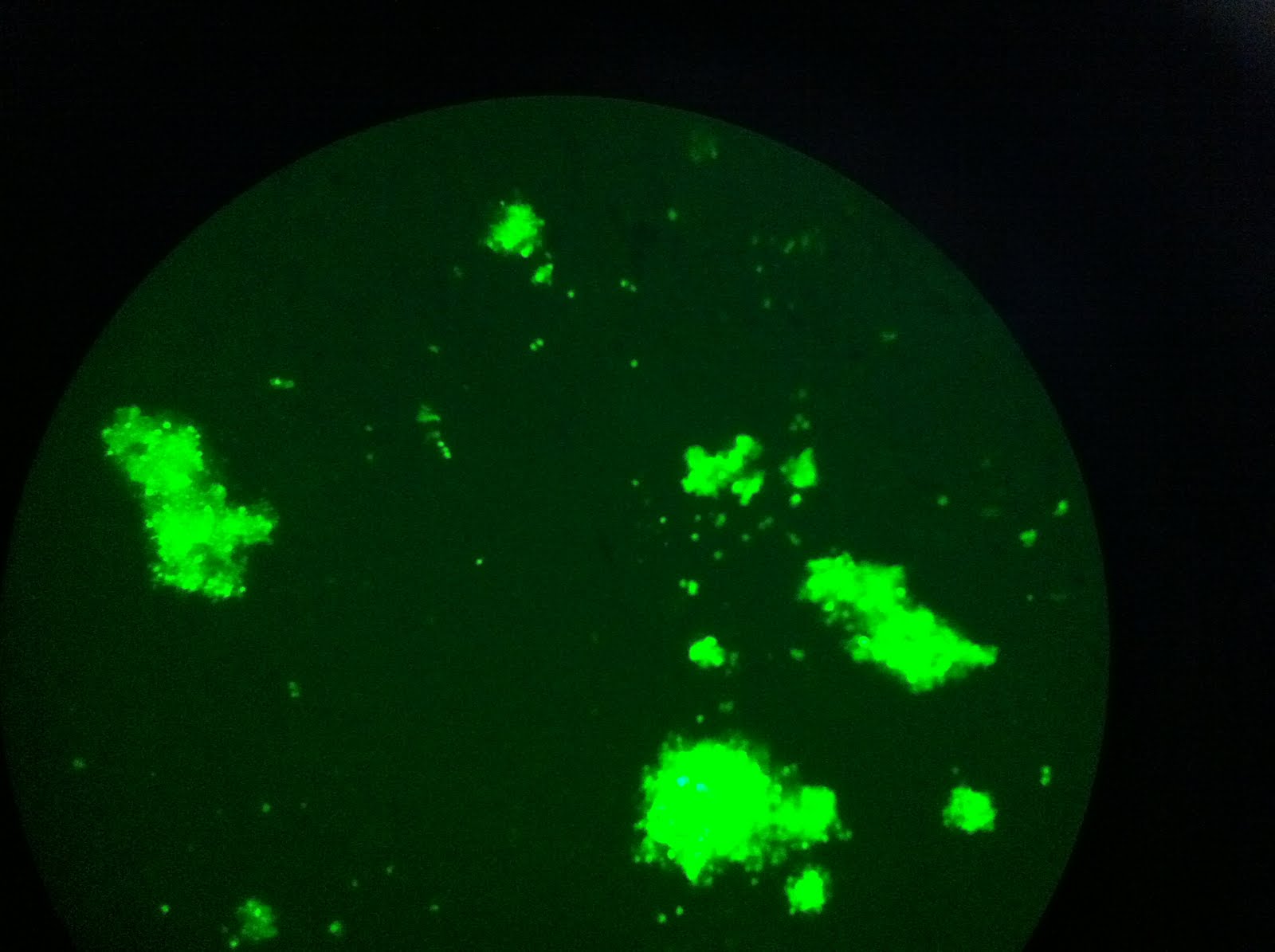 |
| The view through the microscope. So much green! |
 |
| This frame shows some of the variability in the intensities of fluorescence. Denser cell populations will appear brighter. |
 |
| Some clumps of cells are larger than others. |
Sunday, September 4, 2011
The start of the semester & culturing transformed diatoms
 | |
| The backside of Goddard Library, one of my favorite photo spots. |
On Friday, we had our annual student activities fair, where almost every social club on campus from CUFS (Clark U Film Society, which shows movies on Tuesdays and Thursdays in our cinema) to ROCU (Radio of Clark U) is represented by student members. This is a great way for incoming first years to discover all of what Clark has to offer for our ever expanding extracurricular groups.
 |
| A panorama of the student activities fair, as seen from Main St. |
 |
| The fair from the window of Tilton Hall, nearly showing the fair in its entirety. |
Saturday, July 2, 2011
Sup July? SUP SUCCESSFUL TRANSFORMATIONS?
I've been having trouble transforming E. coli cells with my plasmid vector, within which is a small DNA fragment (my NiR terminator) that I will want to restriction cut out. By transforming bacteria with the plasmid vector, I'll make additional copies of the plasmid and be able to freeze and save the plasmid for later use if necessary.
My lab mates and I spent several weeks trying to figure out why our transformations were doing so poorly and why we were receiving such low plasmid yields from transformed bacteria. I myself figured out that one problem was the ampicillin used to make the agar plates upon which we grow our bacteria had degraded over time, and that the antibiotic was not selecting strongly enough to weed out bacteria with plasmids and bacteria without plasmids. This is the reason why we were not getting good plasmid yields and another reason why our bacteria were not growing when transferred from "old" plates to new agar plates with freshly made ampicillin.
We also concluded that the bacteria cells we were transforming were not up to par to yield the results we needed, so we ordered some new transformation kits.
But in order to successfully clone PCR product into a plasmid vector to transform into bacteria, the PCR product needs to be freshly made. In order to get new PCR product, I re-amplified older PCR product in the same reaction I ran before. I ran four different reactions using the PCR DNA in four different DNA concentrations: 1:1, 1:10, 1:100, & 1:1,000 (lanes 2, 3, 4 & 5 in the picture below respectively). This way I can determine which reaction had too much starting DNA and too little. After my reaction, I ran part of it on a gel to see how each reaction went. I definitely got much larger yields in the 1:1 & 1:10 dilutions (there was probably too much DNA even), so I used the second dilution (1:100, lane 4) to clone into the plasmid vector.
Using the vector plasmid, I transformed them into the bacteria and let them grow over night on an agar plate. I then performed a colony screen, which is a PCR reaction using single bacteria colonies to supply the DNA. That PCR reaction yielded the below gel:
What we're seeing in this gel is the molecular ladder at the top and then 10 different colony screen reactions. They're pretty streaky, which is probably because there was a lot of bacterial DNA in each PCR reaction. What I wanted was a single band at around 700 basepairs, which is roughly half way between the two second most right bands on the ladder. As such, lanes 4, 6, 7 & 8 are good candidates for colonies that have my plasmid with the correct insert.
BRB time for the holiday weekend!
My lab mates and I spent several weeks trying to figure out why our transformations were doing so poorly and why we were receiving such low plasmid yields from transformed bacteria. I myself figured out that one problem was the ampicillin used to make the agar plates upon which we grow our bacteria had degraded over time, and that the antibiotic was not selecting strongly enough to weed out bacteria with plasmids and bacteria without plasmids. This is the reason why we were not getting good plasmid yields and another reason why our bacteria were not growing when transferred from "old" plates to new agar plates with freshly made ampicillin.
We also concluded that the bacteria cells we were transforming were not up to par to yield the results we needed, so we ordered some new transformation kits.
But in order to successfully clone PCR product into a plasmid vector to transform into bacteria, the PCR product needs to be freshly made. In order to get new PCR product, I re-amplified older PCR product in the same reaction I ran before. I ran four different reactions using the PCR DNA in four different DNA concentrations: 1:1, 1:10, 1:100, & 1:1,000 (lanes 2, 3, 4 & 5 in the picture below respectively). This way I can determine which reaction had too much starting DNA and too little. After my reaction, I ran part of it on a gel to see how each reaction went. I definitely got much larger yields in the 1:1 & 1:10 dilutions (there was probably too much DNA even), so I used the second dilution (1:100, lane 4) to clone into the plasmid vector.
 |
| I used PCR product from lane 4 to clone into a vector plasmid for the transformation. |
 |
| While this is a slightly messy colony screen gel, several of these colonies should suffice! |
What we're seeing in this gel is the molecular ladder at the top and then 10 different colony screen reactions. They're pretty streaky, which is probably because there was a lot of bacterial DNA in each PCR reaction. What I wanted was a single band at around 700 basepairs, which is roughly half way between the two second most right bands on the ladder. As such, lanes 4, 6, 7 & 8 are good candidates for colonies that have my plasmid with the correct insert.
BRB time for the holiday weekend!
Tuesday, June 14, 2011
It's Transformation Time.
Last time I checked in, my PCR reactions that were supposed to add restriction sites to either end of the nitrite reductase (NiR) terminator region appeared to work and work well. When I ran the PCR reactions on a gel, the amplified DNA bands were really strong and were the correct length.
 This gave me the go ahead to continue the path in isolating & altering the NiR terminator in order to yield a sequence of DNA to fit the final NiR plasmid for my project's experiments.
This gave me the go ahead to continue the path in isolating & altering the NiR terminator in order to yield a sequence of DNA to fit the final NiR plasmid for my project's experiments.
Now I need to insert the NiR terminator into a vector plasmid, transform it into bacteria, and digest the NiR terminator back out of the plasmid to double check that the terminator I got on the gel from my PCR reaction (above) is the correct piece of DNA before I insert it into the NiR plasmid to complete the final NiR plasmid.
We use bacteria to amplify pieces of DNA because of their quick generation times. If you insert a plasmid into bacteria, they will duplicate the plasmid as if it were their own DNA as they grow and divide. A plasmid is a ring of DNA, which is essential for this to work, because bacteria will cut up and destroy any loose pieces of linear DNA. In order to get our NiR terminator to be duplicated by the bacteria, we insert it into a vector plasmid first. The vector plasmid is designed to accept small pieces of DNA from PCR reactions, lock in that piece of DNA within the plasmid. This plasmid can then be transformed into bacteria.
Bacterial transformation is really easy. Once the vector plasmid complete with our PCR DNA is ready, we add the plasmids to specially-altered E. coli cells, incubate the cells on ice for a short time (to lull them into a false sense of security), and then transfer them to a hot water bath (42°C) for thirty seconds. Thirty seconds is all we need for the bacteria cells to panic and scream "WHAT IS HAPPENING TO ME?" This prompts the bacteria, because they are stressed, to take up any DNA in their environment. Well good thing the only DNA in their environment is the plasmid we gave them! Through the heat shock, a large amount of bacteria should have taken up our plasmid. We then grow the bacteria over night while they recuperate, divide exponentially, and make copies of our plasmid. This process is summarized by the cartoon below:

Now I need to insert the NiR terminator into a vector plasmid, transform it into bacteria, and digest the NiR terminator back out of the plasmid to double check that the terminator I got on the gel from my PCR reaction (above) is the correct piece of DNA before I insert it into the NiR plasmid to complete the final NiR plasmid.
We use bacteria to amplify pieces of DNA because of their quick generation times. If you insert a plasmid into bacteria, they will duplicate the plasmid as if it were their own DNA as they grow and divide. A plasmid is a ring of DNA, which is essential for this to work, because bacteria will cut up and destroy any loose pieces of linear DNA. In order to get our NiR terminator to be duplicated by the bacteria, we insert it into a vector plasmid first. The vector plasmid is designed to accept small pieces of DNA from PCR reactions, lock in that piece of DNA within the plasmid. This plasmid can then be transformed into bacteria.
Bacterial transformation is really easy. Once the vector plasmid complete with our PCR DNA is ready, we add the plasmids to specially-altered E. coli cells, incubate the cells on ice for a short time (to lull them into a false sense of security), and then transfer them to a hot water bath (42°C) for thirty seconds. Thirty seconds is all we need for the bacteria cells to panic and scream "WHAT IS HAPPENING TO ME?" This prompts the bacteria, because they are stressed, to take up any DNA in their environment. Well good thing the only DNA in their environment is the plasmid we gave them! Through the heat shock, a large amount of bacteria should have taken up our plasmid. We then grow the bacteria over night while they recuperate, divide exponentially, and make copies of our plasmid. This process is summarized by the cartoon below:
Subscribe to:
Posts (Atom)